

Nugen's Ovation RNA-Seq V2
Nugen's Ovation® RNA-Seq
The Ovation® RNA-Seq System V2 provides a rapid, simple and sensitive whole transcriptome RNA amplification process for preparing amplified cDNA from total RNA or poly(A) selected mRNA from 500pg – 100ng. This robust workflow, based on NuGEN’s proprietary Ribo-SPIA technology, yields several micrograms of cDNA ready for multiple downstream applications including RNA-Seq, qPCR and archival storage for future analyses. When combined with the Ovation® Ultralow V2 Library or Ovation® Rapid Library Systems, a complete RNA-Seq library can be generated in under 7 hours. The product is particularly useful for detection of rare transcripts, viruses and unknown pathogens.
Sample types include total RNA from LCM tissue, fine needle aspirates, sorted cells, liquid biopsy, prokaryotes, viruses and other challenging sample types. Automation enabled on multiple platforms including Agilent Bravo, Beckman Biomek FXP, Perkin Elmer Sciclone, and Hamilton Star/Starlet.
Simple, fast workflow that integrates seamlessly into multiple downstream workflows
- Workflow completed in under 5 hours.
- Micrograms of DNA obtained for downstream applications such as RNA-Seq, qPCR, microarrays and sample archiving.

The cDNA produced by the Ovation® RNA-Seq System V2 may be used as input for NuGEN’s Ovation® library systems, or used with other library construction kits suitable for double-stranded cDNA samples.
Obtain reproducible sequencing results
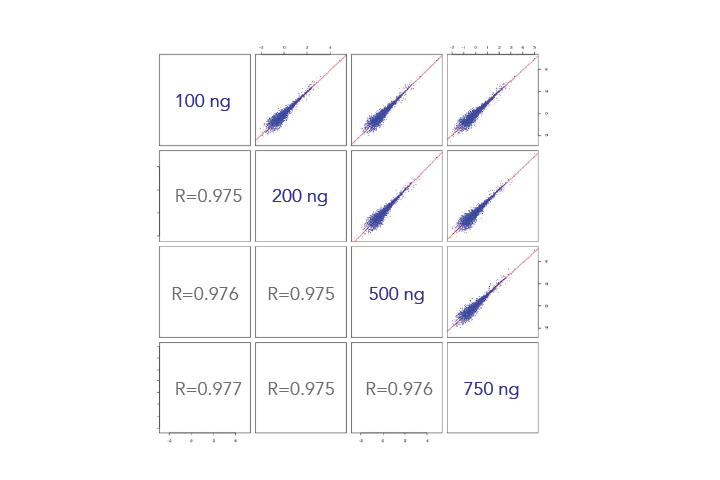
- High concordance seen with all sample input amounts
- Proprietary combination of enzymes and primers allows for preferential priming of non-rRNA sequences with reduced number of reads from rRNA
Total RNA from Human Brain (MAQC B, 2.0 ng) was amplified using either the original Ovation® RNA-Seq System or the Ovation® RNA-Seq System V2 and libraries constructed using the Encore® NGS Multiplex System I. Single-read sequencing results were obtained using the Illumina Genome Analyzer IIx platform with 40 bp reads. The data shown in Figures 4A, B and C show examples of the sequencing read distribution across the indicated transcripts and illustrate the improved exon read coverage with Ovation® RNA-Seq System V2. Results depicted here are for GAPDH gene.
Obtain even exon coverage
- Even distribution of sequencing reads across exons as shown with GAPDH and beta-actin transcripts
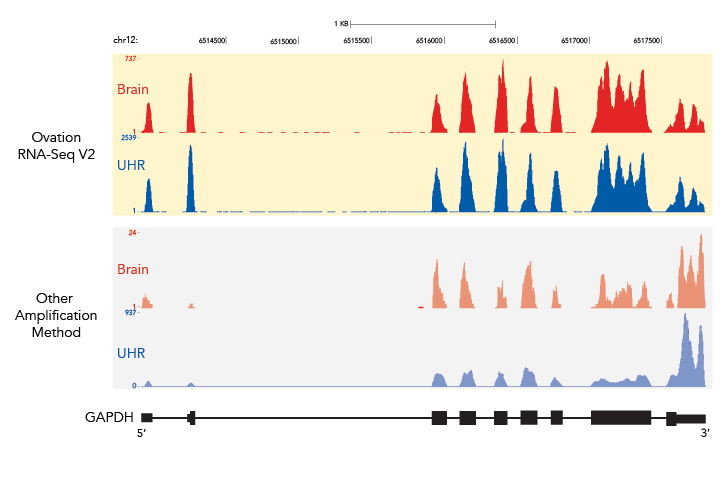
Total RNA from Human Brain (MAQC B, 2.0 ng) was amplified using either the original Ovation® RNA-Seq System or the Ovation® RNA-Seq System V2 and libraries constructed using the Encore® NGS Multiplex System I. Single-read sequencing results were obtained using the Illumina Genome Analyzer IIx platform with 40 bp reads. The data shown in Figures 4A, B and C show examples of the sequencing read distribution across the indicated transcripts and illustrate the improved exon read coverage with Ovation® RNA-Seq System V2. Results depicted here are for GAPDH gene.
Obtain high confidence in data for differential gene expression
- Highly concordant results seen when compared to differential gene expression data from quantitative PCR
- No significant compression of data as evidenced by the slope
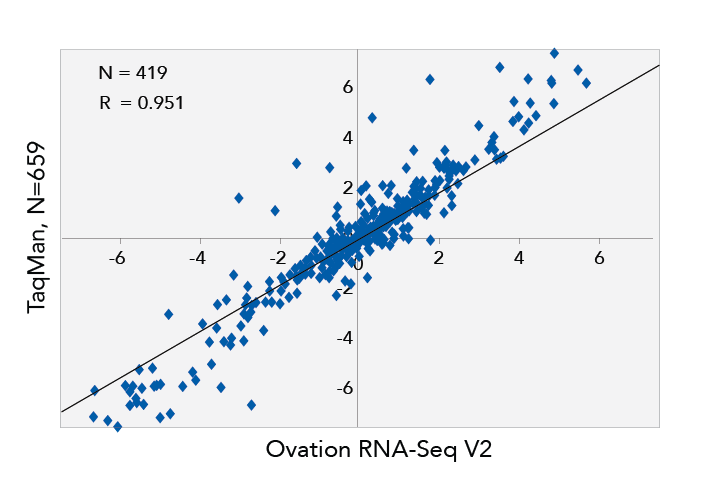
Differences between Log2 transformed expression values for MAQC A and MAQC B samples, RNA-Seq data using 2 ng total RNA as input are plotted on the X axis, qPCR data are plotted on the Y axis. 659 TaqMan probes that uniquely map to the RefSeq annotations used in the RPKM calculations are represented. RPKM stands for Reads Per Kilobase of exon model per Million mapped reads. The RPKM measure of read density reflects the molar concentration of a transcript in the starting sample by normalizing for RNA length and for the total read number in the measurement.